Способ получения гидразина высокой чистоты. Как сделать гидразин в домашних условиях
Чем осадить золото и как это правильно сделать?
В этой статье:
Процесс аффинажа золота, то есть его очищения, проходит в несколько этапов: растворение, фильтрование и осаждение золота. И если вы прошли два начальных этапа очистки драгметалла, у вас наверняка возникнет вопрос, чем осадить золото.
Золотой предмет боле низкой пробы растворяется в царской водке, а далее выпаривается и фильтруется. Процесс достаточно трудоемкий, но многие проводят его дома. Но поскольку это небезопасно, не забывайте о правилах техники безопасности.
И поскольку на выходе двух этапов золото находится в растворенном виде, чтоб извлечь его из раствора, необходимо его осадить. С точки зрения химии, этот процесс называется восстановлением. Поэтому для него нужно использовать вещества, которые имеют восстановительную способность.

Вещества для осаждения золота
Осаждение золота можно проводить с помощью таких реактивов, как:
- Гидразин. Наиболее сильный восстановитель, поэтому использование в домашних условиях опасно. Само вещество обладает неприятным запахом, в качестве восстановителя необходима большая концентрация гидразина. Но в этом случае осадок в виде золота может получиться мелкодисперсным, что затруднит его использование в дальнейшем. Для осаждения лучше использовать сульфат гидразина, чем нитрат гидразина. А еще восстановитель лучше подсыпать в сухом виде, чем подливать в растворе. Способ восстановления золота гидразином довольно сложный для новичков. К тому же гидразин можно использовать для восстановления золота в мелкодисперсных частичках. Вследствие этого процесса получаются коллоидные растворы, в которых золото находится в наночастицах и его можно извлечь путем электрофореза, хоть и сам процесс трудоемкий.
- Щавелевая кислота. Вещество, которое также используется в процессе осаждения. Хотя для первичного проведения процедуры она мало подходит, поскольку обладает меньшими восстановительными способностями. А вот для повторного осаждения можно использовать этот реагент, тогда проба металла будет выше.
- Перекись водорода — это вещество также осаждает золото из растворов. Но для успешности процесса концентрация перекиси должна быть высокой.
- Пиросульфит натрия. Способы также больше подходят для вторичного осаждения металла. Тогда раствор уже более насыщенный, и концентрация азотной кислоты в нем меньше. Пиросульфит следует добавлять в чистом виде, предварительно растворив его в воде без добавления других элементов. Если провести процедуру неправильно можно получить выделение токсичных газов. А если добавить слишком большое количество пиросульфита при таком же большом количестве азотной кислоты в растворе, золото продолжит растворяться и образовывать весьма стойкие комплексы, которые будет сложно отделить в дальнейшем.
- Железный купорос. Осаждение золота железным купоросом — дешевая и надежная процедура. Восстанавливает золото из царской водки или из хлорида золота. Соотношение купороса и раствора колеблется от 2:1 при слабой кислотности до 5:1, чтоб нейтрализовать азотную кислоту. Растворить купорос следует в теплой воде и добавить к нему соляную кислоту. Далее понемногу следует вливать восстановитель в раствор и оставить его на день до осаждения золота. Если вы видите, что реакции не происходит, добавьте еще немного купороса. Способ достаточно простой, но само золото на выходе получается немного грязным. Поэтому методика подходит либо для новичков в этом деле, либо для первичного осаждения драгметалла.
 Железный купорос
Железный купоросЕсть еще много довольно интересных методов для осаждения, они замечательно подойдут для экспериментов. Например, используют винную кислоту, после которой золото выпадает черным осадком. А также осаждают золото сероводородом, оксалатами щелочных металлов. С помощью хлорида сурьмы происходит осаждение золота из его хлорида.
Сульфит натрия в этом процессе также распространенный компонент. Но этот способ восстановления не подойдет для осаждения золота из царской водки, только из его хлорида.
Что не стоит использовать в процессе?
Есть также вещества, с которыми нет смысла проводить эксперименты, потому что они или не дадут абсолютно никакой реакции, или усугубят процесс восстановления. В этот перечень входят:
- Цианид калия. Способ подходит только для восстановления из нейтрального раствора хлорида золота. В остальных случаях реакции не произойдет.
- Гидрат аммиака и карбонат аммония. Вещества не стоит использовать в процессе. Если осаждение и произойдет — высушивать полученное золото опасно для здоровья. В результате процесса получается гремучее золото, которое имеет свойство неожиданно взрываться.
- Нитрат ртути. При осаждении получается мелкодисперсный осадок, а пары, которые возникают в процессе, смертельно опасны.
- Цитраты, тартраты и ацетаты калия не помогут восстановить золото из царской водки.
И хотя в результате всех действий можно получить золото 999 пробы. Восстановление металла — трудоемкий процесс, который не покорится с первого раза новичкам. Важно соблюдать все сроки и пропорции. Поэтому при его выполнении следует обязательно заботиться о собственной безопасности, поскольку цена эксперимента может оказаться действительно высокой.
Гидразин диамид N2h5 — Знаешь как
Бесцветная жидкость с неприятным запахом . Он очень реакционно способен и весьма неустойчив , легко разлагается под воздействием катализаторов , нагревании и воздействию излучений
Получение гидразина .
Его получают окислением мочевины ( карбамид ) CO( Nh3 )2 или аммиака Nh4 с помощью окислителя гипохлорита натрия NaOCl :
Реакция с карбамидом протекает при повышенном давлении и температуре примерно 100 °C в щелочной среде :
t ~100 °C
h3NCONh3 + NaOCl + 2NaOH → N2h5 + h3O + NaCl + Na2CO3
Гидразин получают окислением аммиака при повышенном давлении и температуре реакция протекает в два этапа с образованием хлор амина ,далее хлор амин подвергается реакции с аммиаком , что приводит к образованию гидразина :
Nh4 + NaClO → Nh3Cl + NaOH
Nh3Cl + Nh4 → N2h5 · HCl
Применение
В органической химии для восстановления карбонильной группы альдегидов и кетонов до метиленовой . Реакция идёт через образование гидразонов . Также известны соли гидразина хлорид гидразиния N2H5Cl иногда записывают N2h5 · HCl и сульфат гидразиния N2H6SO4 иногда записывают N2h5 · h3SO4 , последний относят к числу важнейших солей гидразина . Из него в медицине получают некоторые противоопухолевое средство , используют как интенсивный восстановитель в никелировке , в производстве пестицидов и бактерицид для применения в сельском хозяйстве .
Гидразин , а также его соли применяются в качестве восстановителей в получении золота, серебра , платиновых металлов из их солей .
Описание гидразина и его солей .
Гидразин , N2h5 — легковоспламеняющаяся жидкость . Мол. масса 32.05 ; плотность 1008,5 кг/ м3 ; температура кипения 113, 5°C ; плотность пара по воздуху 1, 1 ; диэлектр. пост. 58,5 ; теплота сгорания 14644 кДж/кг ; растворимость в воде не ограничено . Температура вспышки 40°C ; температура самовоспл. 132 °C ; конц. пределы распр. пламени 4,7 — 100%, 62 — 1300 г/ м3. Оксиды железа и чугун сильно катализируют процесс самовоспламенения гидразина , понижая температуру самовоспл. до 23°C . Гидразин очень реакционноспособен и весьма неустойчив ; легко разлагается под влиянием катализаторов , а также при нагревании и воздействии излучений . Склонен к химическому самовозгоранию при контакте с оксидами некоторых металлов ( Cu , Fe , Mo , Cr , Pb , Hg ) или вещества с развитой поверхностью ( уголь , асбест и т.д ) . Пары гидразина , смешанные с разными разбавителями , способны распространять пламя при 104 — 135°C в пределах следующих концентраций : при разбавлении азотом 38 — 100% (об.) ,
Гидразин — гидрат , N2h5 · h3O , легковоспламеняющаяся жидкость. Мол. масса 50,06 ; плотность 1030 кг/м3 ; температура кипения 120°C ; плотность пара по воздуху 1,8 ; растворимость в воде неограниченная . Температура всп. 59°C (о.т) ; т. воспл. 59°C ; температура самовоспламенения 267°C .
Гидразин — сульфат , сульфат гидразина h5N2 · h3SO4 , трудно горючее кристаллическое вещество , сильный восстановитель . Мол. масса 130,13 ; плотность 1378 кг/м³; температура плавления 254°C с разложением ; растворимость в воде 3% ( массе ) при 22°C . Температура самовоспл. 840°C .
znaesh-kak.com
Способ получения гидразина высокой чистоты
Изобретение относится к области процессов химической технологии, а точнее к области технологии гидразиновых соединений. Разработан способ получения гидразина высокой чистоты из гидразин-гидрата и водного гидразина, который включает стадию дегидратации с использованием твердой щелочи, стадию ректификации при пониженном давлении и стадию фракционной кристаллизации, причем стадию дегидратации проводят в режиме интенсивного перемешивания и эмульгирования образующихся жидких фаз; стадию ректификации проводят в непрерывном режиме после отделения основной части щелочи-едкого натра до остаточного содержания не более 0,5 мас.% методом охлаждения и отстаивания, давление при ректификации поддерживают в диапазоне 50-200 мм рт.ст., при числе теоретических тарелок колонны не менее 6, флегмовом числе 2-4; а стадию кристаллизации гидразина проводят в условиях перемешивания и удаления кристаллов со стенок кристаллизатора с переводом процесса кристаллизации в объем аппарата, до степени кристаллизации не более 80% со скоростью кристаллизации не выше 30 кг/час на 1 м2 площади охлаждаемой поверхности аппарата. Техническим результатом изобретения является уменьшение энергозатрат и повышение безопасности производства. 2 з.п. ф-лы, 2 ил., 1 табл., 11 пр.
Изобретение относится к области процессов химической технологии, а точнее к области технологии гидразиновых соединений. Гидразин (или диамид) Nh3-Nh3, Безводный гидразин при нормальных условиях представляет собой бесцветную дымящую на воздухе жидкость. Гидразин смешивается с водой в любых соотношениях. Состав, содержащий 50 моль % гидразина и 50 моль % воды, называется гидразин-гидрат. Растворение гидразина в воде - процесс экзотермический [Н.В. Коровин. Гидразин, М., «Химия», 1980].
Гидразином высокой чистоты считают продукт, содержащий не менее 99% основного вещества (гидразина), не более 0,5% воды, не более 0,3% аммиака и не более 0,001% примесей, образующих нелетучий остаток. Выпускают ряд марок гидразина высокой чистоты, в том числе гидразин особой чистоты с содержанием основного вещества (гидразина) не менее 99,5%, воды не более 0,4%, аммиака не более 0,1% и не более 0,0002% примесей, образующих нелетучий остаток, что соответствует марке ГХЧ-2 СТО [СТО 40059405-09-2009, СМК, Гидразин химически чистый. Технические условия].
В настоящее время в промышленно развитых странах возрастает потребность в гидразине высокой чистоты. Гидразин используется в ракетно-космической отрасли, фармацевтической и других отраслях промышленности.
Сырьем для получения гидразина высокой чистоты является гидразин-гидрат. Технология получения гидразина из гидразин-гидрата состоит из стадий дегидратации гидрата и последующей очистки сырца гидразина до заданной чистоты. Известно большое число вариантов технологии дегидратации и выделения гидразина. Например, в работе [Патент США 4804442, МПК B01D 3/10, опубл. 14.02.1989] описана технология получения гидразина высокой чистоты из гидразин-гидрата, состоящая из двух стадий:
- дегидратации твердой гранулированной щелочью (едким натром),
- ректификации в мягких условиях при температуре куба колонны 30-40°С и давлении от 3 до 15 мм рт. ст.
В результате получается гидразин с чистотой не менее 99,7%. Применение этого метода для удаления примесей, образующих нелетучий остаток, малоэффективно и требует применения ректификации с большим числом тарелок. Применение ректификации в качестве единственного метода очистки приводит к повышению пожаро- и взрывоопасности, потерям продукта и увеличению экологических проблем.
Описан способ [Патент США 5035775, МПК B01D 3/10, опубл. 30.07.1991 г.], позволяющий получать гидразин, свободный от органических примесей. Защищаются мольные отношения гидроксида щелочного металла к воде на стадии дегидратации (от 0,15 до 0,5), а также условия двухстадийной дистилляции: давление менее 60 мм рт.ст., температура от 10 до 50°С. Недостатком метода являются значительные потери гидразина в составе кубовой жидкости и высокая опасность процесса из-за периодичности процесса и длительного пребывания больших объемов гидразина в зоне повышенных температур. Метод представляет интерес только как лабораторный.
В патенте [EP 0226686 МПК C01B 21/16, опубл. 20.12.85] описана технология очистки гидразина, полученного путем дегидратации гидразингидрата щелочью. Очистка производится путем отделения щелочи (едкого натра) на ионообменной колонке и многократной перекристаллизации полученного гидразина в длинной трубе методом зонной плавки. Показана возможность получения гидразина с концентрацией, превышающей 99%, после нескольких операций перекристаллизации. Недостатком данного метода является то, что применение ионообменных смол сопряжено с необходимостью их регенерации и образования большого количества растворов солей, загрязненных гидразином, а метод зонной плавки имеет низкую производительность и требует многократного повторения операций плавления и кристаллизации. Данный метод целесообразно применять только в лабораторной практике.
В работе [A.J. Bougine, R. Tenu, J. Berthet, A. Dhenain, H. Delalu, JJ. Counous. Synthesis and Extraction of Anhydrous High Purity Hydrazine (HPH) from the Hydrazine Hydrate, 19th ICCT 2006/61st CalCon (IUPC International Conference on Chemical Thermodynamics / Calorimetry Conference). 30/07-04/08/2006, Boulder (Colorado, United States)] описана технология синтеза и очистки гидразина, обеспечивающая получение гидразина с содержанием углерода не более 30 ppm. Исходным продуктом для получения гидразина является гидразин-гидрат, полученный методом Рашига. В работе описаны стадии дистилляции азеотропа гидразина с последующей дегидратацией в присутствии щелочи (NaOH) и фракционной кристаллизации при пониженной температуре. Этот метод ограничен производством гидразина по методу Рашига, поскольку исключена стадия ректификации гидразина-сырца, и не позволяет использовать гидразин-гидрат, полученный кетазиновыми методами.
Некоторые результаты по очистке гидразина от примесей методом направленной кристаллизации приведены в работе [Л.В. Литвинова, К.П. Мищенко, В.В. Кущенко. Получение чистого гидразина методом направленной кристаллизации, ЖПХ, т. LI, №11, 1978 г, с. 2610-2691]. Под направленной кристаллизацией понимают большую группу методов, основанных на направленном отводе тепла от границы раздела фаз, вызывающем в свою очередь направленное передвижение фронта кристаллизации вдоль очищаемого образца. Это передвижение (чаще всего с постоянной скоростью) осуществляется принудительно путем постепенного перемещения зон охлаждения и нагрева.
Результаты работы позволяют предвидеть степень очистки гидразина при проведении кристаллизации со скоростью 20 мм/час при температуре хладагента минус 50°С, в условиях интенсивного перемешивания. Для получения чистого гидразина рекомендовано многократное повторение операции кристаллизации со стадиями зонной плавки, что нецелесообразно при промышленном использовании способа.
Процесс очистки гидразина от воды, анилина и других примесей с многократной кристаллизацией большей части гидразина, находящегося в производственной установке, является основой технологии очистки гидразина, приведенной в работе [W.B. Schuler, T.J. Pharo and C.A. Hall, The removal of impurities from control of contamination caused by rocket engine exhaust. AIAA/SAE 8th Joint Propulsion Specialist Conference, New Orleans, Louisiana - November 29-December 1, 1972]. Гидразин, подлежащий очистке, циркулирует через вертикальную трубку, охлаждается и частично замораживается слоем на стенке трубки, при этом жидкая фаза перемешивается в процессе циркуляции. В каждом цикле замораживания рекомендуется вести процесс до кристаллизации 90% гидразина, загруженного в установку. Затем отделяют маточный раствор и производят плавление кристаллического слоя с разделением на фракции. Для достижения высокой степени очистки продукта требуется многократное повторение цикла замораживание - плавление.
В результате применение этого метода приводит к большому расходу энергии и повышению себестоимости товарной продукции.
В процессе исследования методов перекристаллизации установлено, что одним из наиболее эффективных методов очистки является метод противоточной фракционной кристаллизации [Н.И. Гельперин, Г.А. Носов, Основы техники кристаллизации расплавов; Москва, Химия, 1975], который заключается в том, что кристаллизацию и плавление проводят в аппарате колонного типа, причем верхнюю часть колонны охлаждают, и там происходит образование кристаллов, которые при помощи шнека или самопроизвольно, за счет разности плотностей твердого продукта и расплава перемещаются в нижнюю часть аппарата, где нагреваются и плавятся.
Часть расплава выводится из нижней части колонны и представляет собой очищенный продукт, но большая часть расплава движется вверх, навстречу потоку кристаллов. При обтекании кристаллов восходящим потоком расплава происходит промывка кристаллического материала от продуктов, окклюдированных в процессе образования и роста кристаллов, и перемещение примесей вверх, в зону кристаллизации. Из верхней части аппарата производится отбор маточного раствора с высоким содержанием примесей. Ввод исходного продукта осуществляется в среднюю или верхнюю часть аппарата.
Таким образом, за время пребывания некоторой порции вещества в колонне происходит многократная кристаллизация и расплавление этого вещества и возможна значительно более полная очистка его от примесей. Примеры применения метода противоточной фракционной кристаллизации для производства и очистки гидразина не известны.
Наиболее близкой по технической сущности к предлагаемому изобретению является технология получения гидразина высокой чистоты, описанная P. Kletzkine, Н. Papenberg, О. Fischer, [European production of high purity hydrazine, R. Cohen-Adad. AIAA-90-2320]. В работе рассмотрены вопросы дегидратации гидразин-гидрата, дистилляции гидразина-сырца, очистки гидразина методами кристаллизации. Подробно рассмотрена технология очистки гидразина, основанная на методе фракционного плавления. Рассмотрена технология получения гидразина высокой чистоты из гидразина, полученного путем азеотропной ректификации с использованием анилина. Основой технологии является многостадийная перекристаллизация с промывкой осадка фракциями гидразина, полученными на последующих стадиях процесса с передачей маточного и промывочного растворов на предыдущие стадии производства.
К недостаткам описанного способа получения гидразина по прототипу относится то, что допускается очистка непосредственно гидразина-сырца, минуя стадию дистилляции (ректификации). При проведении дегидратации гидразин-гидрата твердой щелочью получаемый гидразин-сырец содержит остаточную щелочь и обладает цветностью.
Для получения гидразина высших сортов, например, гидразина особой чистоты, из этого гидразина-сырца требуется разработка технологии, включающей применение методов, связанных с перегонкой гидразина, а также методов перекристаллизации. Рассматриваемая в прототипе технология перекристаллизации представляет собой разновидность метода фракционного плавления, является периодической. При однократном выполнении комплекса операций перекристаллизации образуется продукт недостаточной чистоты. Это объясняется тем, что в процессе кристаллизации слой гидразина захватывает в себя около 20% маточного раствора. В дальнейшем эта жидкая фаза дополнительно охлаждается и частично кристаллизуется со всеми примесями, которые в ней содержатся. При однократном проведении кристаллизации полученный продукт содержит загрязнения, что снижает качество товарного продукта и требует увеличения числа циклов перекристаллизации. Многократная перекристаллизация усложняет технологию и повышает энергозатраты.
Получение товарного гидразина высокой чистоты является комплексной задачей из-за наличия примесей, различных по своей физико-химической природе, что зависит от метода получения гидразина-сырца. Для каждой примеси требуется разработка оптимального приема очистки, с помощью которого наиболее эффективно эту примесь удаляют.
Так, минеральный нелетучий остаток сложно удалить методами дистилляции и ректификации до требуемых показателей, в то время как вода при ректификации, в основном, остается в кубе колонны. Кроме идентифицированных примесей в гидразине-сырце, полученном путем щелочной дегидратации, присутствуют микропримеси, обуславливающие цветность продукта. Эти примеси эффективно удаляют методом ректификации.
Основной целью при разработке эффективной технологии производства особо чистого гидразина является оптимизация технологии и выбор последовательности стадий получения и очистки гидразина для обеспечения стабильно высокого качества продукции. Необходимо осуществление безопасного ведения процесса при низком энергопотреблении.
Задачей, стоящей перед авторами данного изобретения, является разработка комплекса технических решений в области технологии получения и очистки гидразина, применение которых приводит к получению гидразина высокой чистоты и позволит минимизировать пребывание продукта в зоне повышенных температур. Задачей является также уменьшение энергозатрат и повышение безопасности производства.
Поставленная задача достигается за счет проведения стадий дегидратации, ректификации и кристаллизации в отработанных авторами оптимальных условиях. Так, стадию дегидратации проводят в режиме, с одной стороны, интенсивного массопереноса в зоне контакта гранул щелочи и гидразин-гидрата, а с другой стороны, эмульгирования возникающей при реакции гидразиновой фазы в водно-щелочной фазе. Стадию ректификации проводят в непрерывном режиме, стадию перекристаллизации - в периодическом режиме с промывкой кристаллического слоя или в непрерывном противоточном режиме фракционной кристаллизации. При этом на каждой из этих стадий применяются новые технические решения.
Сущность изобретения состоит в том, что разработан способ получения гидразина высокой чистоты из гидразин-гидрата и водного гидразина, включающий стадию дегидратации с использованием твердой щелочи, стадию ректификации при пониженном давлении и стадию фракционной кристаллизации, отличающийся тем, что
- стадию дегидратации проводят, подавая реагенты в мольном отношении едкий натр: гидразингидрат 0,8-1,8 при температуре 60-80°С в режиме интенсивного перемешивания и эмульгирования образующихся жидких фаз;
- стадию ректификации проводят в непрерывном режиме после отделения основной части щелочи (едкого натра) до остаточного содержания не более 0,5 мас. % методом охлаждения и отстаивания, причем давление при ректификации поддерживают в диапазоне 50-200 мм рт.ст., при числе теоретических тарелок колонны не менее 6, флегмовом числе 2-4;
- стадию кристаллизации гидразина проводят в условиях перемешивания и удаления кристаллов со стенок кристаллизатора с переводом процесса кристаллизации в объем аппарата, до степени кристаллизации не более 80%, со скоростью кристаллизации не выше 30 кг/час на 1 м2 площади охлаждаемой поверхности аппарата;
- интенсивное перемешивание на стадии дегидратации осуществляют резонансно-пульсационным способом со скоростью струи 3-7 м/с и времени импульсного воздействия 0,2-0,7 с;
- стадию кристаллизации проводят в условиях противоточной фракционной кристаллизации при соотношении потоков кристаллов гидразина и гидразина высокой чистоты не менее 3.
Способ осуществляют, проводя стадию дегидратации при интенсивном перемешивании с использованием механических перемешивающих устройств (мешалок), либо путем создания струйных течений в местах контакта жидкой фазы с гранулированной щелочью. Скорость струйных течений от 3 до 7 м/сек, что приводит к интенсивному перемешиванию жидкой фазы (гидразина и гидразин-гидрата) с гранулами щелочи (едкого натра) при соотношении реагентов едкого натра и гидразин-гидрата 0,8-1,8 при температуре 60-80°С, в результате чего происходит эмульгирование двух образующихся жидких фаз: гидразиновой и щелочной, которые после отстаивания разделяются методом декантации при температуре синтеза.
Наиболее эффективно и экономично обе эти задачи можно решить путем применения резонансно-пульсационного метода [Островский Г.М., Малышев П.А., Аксенова Е.Г., ТОХТ, 1990, т. 24, №6, с. 835; Абиев Р.Ш., Аксенова Е.Г., Островский Г.М., Химическая промышленность, 1994, №11, с. 44], при котором за счет воздействия на реакционную массу пульсирующих струй жидкости проходят процессы растворения щелочи и образования эмульсии. Для этого в аппарате для проведения процесса дегидратации (поз. 1, Фигура 1) устанавливают центральную трубу (поз. 2 Фигура 1), в которой имеется определенное число отверстий. В этой системе сообщающихся сосудов организован колебательный процесс с пульсацией давления и уровней в центральной трубе и остальном объеме аппарата. Пульсации создаются пульсатором (поз. 3, Фигура 1) в виде мембраны или сильфона с приводом от электродвигателя. Созданная таким способом система имеет резонансную частоту колебаний, при которой колебания уровня в центральной трубе достигают максимума. В резонансном режиме струи, бьющие из сопел, достигают максимально возможных скоростей, а их воздействие на процессы растворения гранул щелочи и эмульгирования двух жидких фаз максимально. Следует отметить, что для установок промышленного масштаба подбор или разработка перемешивающего устройства традиционного типа для организации процесса растворения гранулированной щелочи и процесса эмульгирования представляет собой сложную техническую задачу, которая осложняется для гидразина как высокотоксичного, взрывоопасного продукта. Поэтому резонансно-пульсационный метод является на сегодня единственным простым способом решения поставленной задачи.
Полученный таким образом гидразин-сырец (гидразиновая фаза), содержащий едкий натр до 2% и воду до 6%, охлаждают до температуры от 0 до 10°С, выдерживают при этих условиях в течение не менее 10 часов, декантируют гидразин-сырец с содержанием едкого натра не более 0,5% и передают на стадию ректификации.
Ректификацию гидразина-сырца проводят в колонне, имеющей относительно небольшое число теоретических тарелок (но не менее 6), при небольших флегмовых числах (2-4) при пониженном давлении (50-200 мм рт. ст.) в непрерывном режиме, при котором время пребывания гидразиновых смесей в области высоких температур минимально. Применение колонны с числом теоретических тарелок более 6 приводит к повышению качества товарной продукции. Увеличение числа теоретических тарелок более 15 возможно, однако почти не влияет на эффективность дополнительной очистки гидразина и поэтому нецелесообразно из технико-экономических соображений.
На стадии ректификации отделяется основная часть примесей и получается бесцветный гидразин, содержащий менее 1% примесей (например, воды, аммиака и примесей, создающих нелетучий остаток), которые отделяются на стадии перекристаллизации.
На стадию перекристаллизации поступает гидразин-ректификат с предыдущей стадии или гидразин, сходный по составу с гидразином-ректификатом, но полученный из другого источника.
Процесс перекристаллизации проводят так, чтобы образование кристаллов происходило, в основном, в объеме аппарата так, чтобы получался осадок, который можно было бы легко отделить от маточного раствора (например, путем фильтрации) и при необходимости промыть. Для повышения производительности кристаллизатора следует обеспечить интенсивную теплоотдачу от кристаллизующейся массы к стенкам аппарата, а также не допускать образования толстого слоя кристаллического продукта на стенках кристаллизатора и, кроме того, обеспечить появление зародышей твердой фазы в зоне кристаллизации. Экспериментально скорость кристаллизации определяют с учетом времени кристаллизации по количеству маточного раствора и очищенного гидразина, полученному в опыте.
Дополнительный положительный эффект можно получить, если использовать на стадии перекристаллизации гидразина метод противоточной фракционной кристаллизации, реализованный, например, в колонном аппарате, состоящем из трех секций. Ввод исходного продукта осуществляют в среднюю секцию, вывод очищенного продукта - из нижней секции, а вывод маточного раствора - из верхней секции. При этом верхняя секция выполняет роль кристаллизатора, а нижняя секция - роль плавителя очищенного кристаллического продукта.
Значение кратности циркуляции гидразина в аппарате, то есть отношения потока кристаллов к потоку очищенного гидразина, отбираемого из аппарата, следует поддерживать не менее 3. Измерение потока кристаллов гидразина и соотношения потоков кристаллов гидразина и гидразина высокой чистоты представляет собой весьма сложную задачу. Поэтому поток кристаллического гидразина, выходящий из верхней секции колонны, определяют расчетным путем по тепловому потоку, отводимому из верхней секции колонны. Тепловой поток, отводимый из колонны, пропорционален разности температур кристаллизующейся массы и хладагента, площади поверхности теплоотдачи и коэффициенту теплоотдачи. Разность температур определяют экспериментально, площадь теплоотдачи - заданный конструктивный параметр, а коэффициент теплоотдачи определяют по известным эмпирическим зависимостям, опубликованным в литературе для аналогичных конструкций [например, Ф. Стренк, Перемешивание и аппараты с мешалками, стр. 275, Изд. «Химия» Лен. отд., 1975].
Кроме того, рассчитан коэффициент теплоотдачи от стенки аппарата к хладагенту, который равен примерно 3000 Вт/м2·К (градусы Кельвина). Поэтому лимитирующей стадией является теплопередача от кристаллизующейся жидкости к стенке аппарата, а суммарный коэффициент теплоотдачи в данном случае равен 250-300 Вт/м2·К. Для всех примеров, приведенных ниже, площадь теплоотдачи составляет 0,07 м2. Поток кристаллического гидразина, выходящий из верхней секции колонны на единицу площади поверхности теплообмена, представляет собой скорость кристаллизации в аппаратах такого типа.
Общий порядок осуществления стадий представлен на Фигуре 2, где показано, что два исходных потока: гидразин-гидрат и едкий натр подают на стадию дегидратации гидразингидрата (поз. 4), откуда гидразин-сырец передают на стадию ректификации (поз. 5). Со стадии ректификации выводят раствор щелочи и направляют на утилизацию. Гидразин-ректификат после стадии ректификации передают на стадию перекристаллизации (поз. 6), откуда осуществляют возврат обводненного гидразина на ректификацию, а очищенный гидразин выводят из процесса.
ПРИМЕРЫ КОНКРЕТНОГО ИСПОЛНЕНИЯ
Принципиальная схема установки для получения гидразина высокой чистоты приведена на фиг. 2. Принципиальная схема стадии дегидратации гидразин-гидрата представлена на фиг 1. Все примеры, приведенные ниже, выполнены на этой установке, созданной в соответствии с этими схемами.
Пример 1. В реактор дегидратации гидразин-гидрата объемом 2 л загружают щелочь (едкий натр) в количестве 1 кг и гидразин-гидрат в количестве 0,8 кг, что соответствует мольному соотношению 1,56.
Реагенты нагревают в режиме перемешивания со скоростью вращения мешалки 500 об/мин, при температуре 80°С и поддерживают режим перемешивания в течение двух часов. Перемешивание осуществляют при помощи быстроходного перемешивающего устройства (мешалки лопастного типа). Затем выдерживают реакционную массу при температуре 80°С без перемешивания в течение еще двух часов. Реакционную массу отстаивают до образования двух слоев, которые разделяют методом декантации. Верхний гидразиновый слой в количестве 0,45 кг охлаждают, отстаивают и подвергают ректификации в непрерывном режиме на колонне, которая по разделительной способности эквивалентна модели с 15 теоретическими тарелками при флегмовом числе 3. Полученный после ректификации гидразин - ректификат загружают в кристаллизатор. Кристаллизатор представляет собой аппарат с донным фильтром объемом в один литр, снабженный перемешивающим устройством якорного типа. Кристаллизатор охлаждают со скоростью примерно 0,2°С/мин. Процесс кристаллизации продолжают при работающем перемешивающем устройстве в течение нескольких часов со скоростью ~10 кг/час·м2. После этого проводят слив маточного раствора, промывку кристаллического слоя очищенным гидразином, расплавление кристаллического гидразина и слив жидкости в сборник готового продукта. Степень кристаллизации, то есть доля кристаллической фазы в гидразине в этом опыте составляет величину 70%. Результаты опытов, характеризующие качество очищенного гидразина, сведены в таблицу 1.
Пример 2. Технологические параметры этого примера, в основном, соответствуют примеру 1, но в отличие от предыдущего примера перемешивание реакционной массы на стадии дегидратации гидразин-гидрата проводят с использованием резонансно-пульсационного метода перемешивания в соответствии со схемой, представленной на фиг. 1. Резонансно-пульсационное воздействие приводит к перемещению отдельных гранул щелочи и быстрому образованию эмульсии белого цвета, которая, однако, позволяет в дальнейшем выявить перемещения жидкости в аппарате в виде отдельных струйных течений. По полученным результатам пример 2 аналогичен примеру 1.
Пример 3. Условия, в которых осуществляли получение гидразина в этом примере, аналогичны примеру 2, но отличие заключается в том, что перекристаллизацию гидразина проводят в кристаллизационной колонне, работающей в режиме противоточной фракционной кристаллизации.
Для настоящего примера разность температур кристаллизующейся массы и хладагента равна 8°С, суммарный коэффициент теплоотдачи 300 Вт/м2·К, а тепловой поток от кристаллизатора равен 168 Вт. Поток кристаллического гидразина, образующийся в кристаллизаторе, составляет 0,0133 моль/с или 1,5 кг/час. Отбор очищенного гидразина составляет 0,25 кг/час, а соотношение потоков кристаллов гидразина и гидразина высокой чистоты равно 6. В результате такого сочетания технологических параметров достигнуто качество очищенного гидразина, соответствующее особо чистому гидразину.
Пример 4. Сопоставительный
Условия этого примера отличаются от примера 3 тем, что в этом примере отсутствует стадия ректификации. При такой технологии получения гидразина ухудшается качество товарного продукта и наблюдается появление цветности - очищенный гидразин имеет светло-коричневый цвет.
Пример 5
Этот пример показывает влияние режима работы кристаллизационной колонны, а именно, соотношения потоков кристаллов гидразина и гидразина высокой чистоты на результаты очистки гидразина.
В данном случае путем уменьшения разности температур кристаллизатора и хладагента снижен поток кристаллического гидразина и увеличен отбор гидразина очищенного. В результате отношение этих потоков составило около 2, что привело к повышению концентрации примесей в очищенном гидразине.
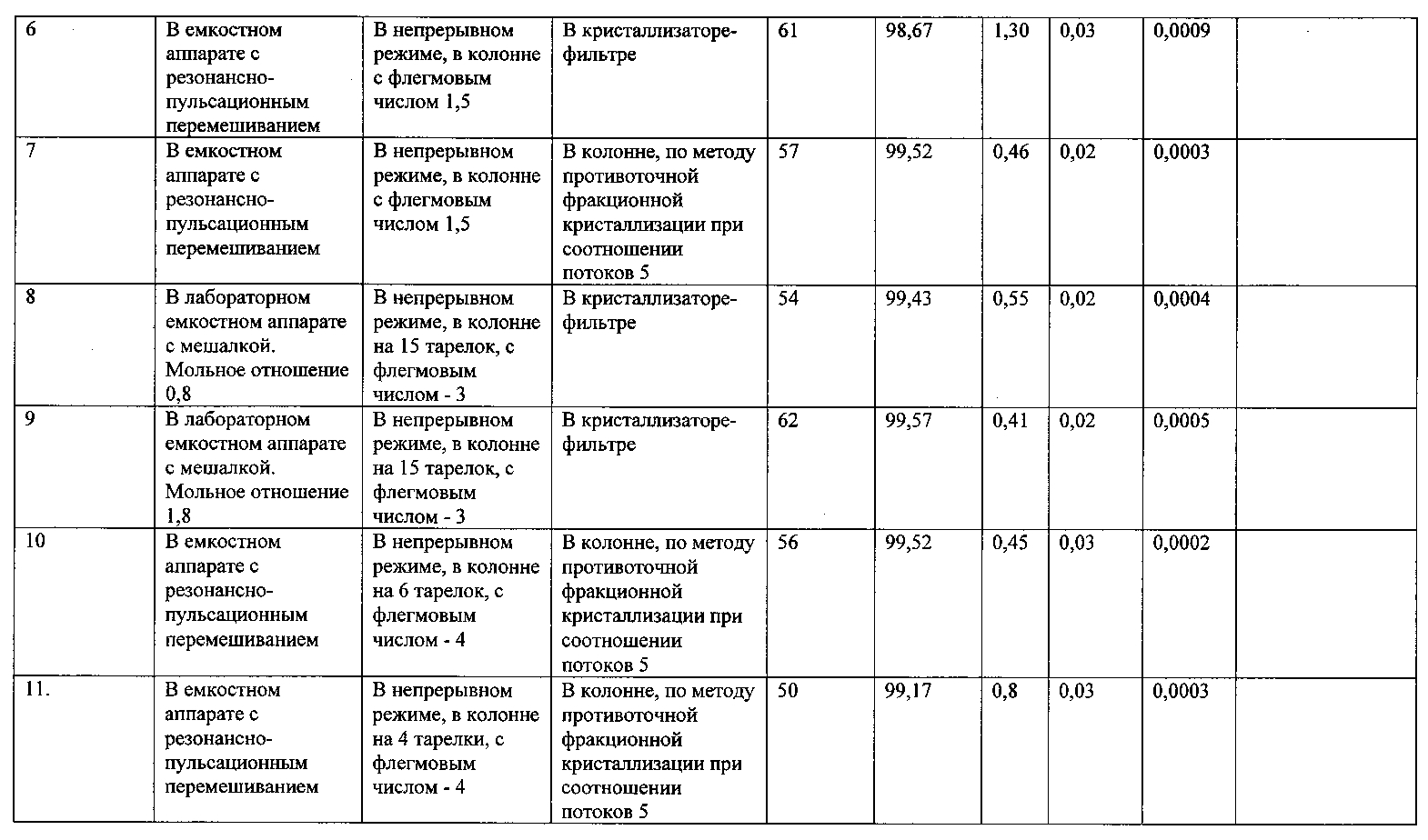
Пример 6. Пример демонстрирует влияние флегмового числа ректификационной колонны на процесс получения и очистки гидразина. Значительное уменьшение флегмового числа (до 1,5) заметно ухудшает результаты работы производства в целом.
Пример 7. Этот пример отличается от предыдущего тем, что на стадии кристаллизации применяется более эффективный метод: метод противоточной фракционной кристаллизации. Поэтому по сравнению с предыдущим примером наблюдается улучшение качества полученного гидразина, однако, этого недостаточно для решения поставленной задачи.
Пример 8. В реактор дегидратации гидразин-гидрата объемом 2 л загружают щелочь (едкий натр) в количестве 0,49 кг и гидразин-гидрат в количестве 0,8 кг, что соответствует мольному соотношению 0,8. Получили верхний гидразиновый слой в количестве 0,4 кг. Остальные операции проводили в соответствии с примером 1. В результате наблюдается некоторое снижение выхода очищенного гидразина при сохранении качества на прежнем уровне.
Пример 9. Этот пример отличается от предыдущего большим избытком щелочи (едкого натра) по отношению к гидразин-гидрату. Щелочи (едкого натра) было взято 1,15 кг, а гидразин-гидрата 0,8 кг (1,8 моль едкого натра на моль гидразин-гидрата). Результаты этого опыта близки к примеру 1, однако дальнейшее увеличение количества щелочи нецелесообразно, так как приводит к неоправданному удорожанию товарного продукта и увеличивает количество отходов.
Пример 10. Этот пример отличается от примера 3 использованием на стадии ректификации колонны с 6 теоретическими тарелками и с большим флегмовым числом, равным 4. Полученный продукт при том же выходе незначительно отличается от примера 3.
Пример 11.Этот пример отличается от примера 3 использованием на стадии ректификации колонны с меньшим числом теоретических тарелок - 4 и большим флегмовым числом 4. Полученный продукт при том же выходе имеет повышенное содержание воды и нелетучего остатка.
Таким образом, решена задача, стоящая перед авторами данного изобретения: разработано техническое решение в области технологии получения и очистки гидразина, применение которых приводит к получению гидразина высокой чистоты и позволит минимизировать пребывание продукта в зоне повышенных температур, достигнуто уменьшение энергозатрат и повышение безопасности производства.

1. Способ получения гидразина высокой чистоты из гидразин-гидрата и водного гидразина, включающий стадию дегидратации с использованием твердой щелочи, стадию ректификации при пониженном давлении и стадию фракционной кристаллизации, отличающийся тем, что- стадию дегидратации проводят, подавая реагенты в мольном отношении едкого натра и гидразин-гидрата от 0,8-1,8 при температуре 60-80°С в режиме интенсивного перемешивания и эмульгирования образующихся жидких фаз;- стадию ректификации проводят в непрерывном режиме после отделения основной части щелочи - едкого натра до остаточного содержания не более 0,5 мас.% методом охлаждения и отстаивания, причем давление при ректификации поддерживают в диапазоне 50 - 200 мм рт.ст. при числе теоретических тарелок колонны не менее 6, флегмовом числе 2-4;- стадию кристаллизации гидразина проводят в условиях перемешивания и удаления со стенок кристаллов с переводом процесса кристаллизации в объем аппарата до степени кристаллизации не более 80%, со скоростью кристаллизации не выше 30 кг/час на 1 м2 площади охлаждаемой поверхности аппарата.
2. Способ по п.1, отличающийся тем, что интенсивное перемешивание осуществляют резонансно-пульсационным способом со скоростью струи 3-7 м/с и временем импульсного воздействия 0,2-0,7 с.
3. Способ по п.1, отличающийся тем, что стадию кристаллизации проводят в условиях противоточной фракционной кристаллизации при соотношении потоков кристаллов гидразина и гидразина высокой чистоты не менее 3.
www.findpatent.ru
Гидразина безводного получение - Справочник химика 21
| Рис. 198. Прибор для получения небольших количеств безводного гидразина из N2H5 1. |  |
Ниже приводятся способы для получения 10—50 г безводного гидразина (содержание около 99,5 масс. %). Большие количества (100—500 г) получают по несколько иным методикам, которые, однако, часто требуют затраты значительно большего времени и более сложной аппаратуры, и поэтому иа них даются только ссылки. Несмотря на то что приведенные способы считаются безопасными, на всякий случай следует отгораживать аппаратуру защитным экраном. [c.491]
В книге описаны реакции, приводяш,ие к образованию гидразина. Особое внимание уделено рассмотрению процесса, лежащего в основе промышленного метода получения гидразина окислением аммиака гипохлоритом (синтез Рашига). При помощи этого метода получается сравнительно разбавленный раствор гидразина. Поэтому в книге подробно рассмотрены способы концентрирования разбавленных растворов и методы получения безводного гидразина. Далее авторы описывают физические, химические и термохимические свойства безводного гидразина, его физиологическое действие, методы качественного и количественного анализа гидразина, а также возможности его практического применения. Специальное место уделено описанию простых и двойных солей гидразина и его координационных соединений. [c.3]
Молекулы гидразина в жидком состоянии связаны между собой водородными связями [79], в парах он мономолекулярен [80]. Гидразин образует азеотроппую смесь с водой, которая кипит примерно при такой же температуре, как безводный гидразин. Для получения безводного растворителя в качестве дегидратирующего средства можно использовать окись бария. Кроме того, благодаря нерастворимости сульфата аммония в гидразине безводный растворитель можно получить по реакции солей гидразипия с жидким аммиаком [81, 82] [c.65]
Сульфат гидразина был получен при поглощении безводного азида водорода концентрированной серной кислотой [38]. Механизм этой реакции не был точно выяснен однако и в этом случае можно сделать предпаложениг о промежуточном образовании радикала НМ. [c.22]
Обычно применяемый способ заключается в обработке метилового или этилового эфира кислоты гидразином. В большинстве случаев пользуются не безводным гидразином, а технически доступным 85 /о-ным водным гидразингидратом. Образование гидразидов из сложных эфиров часто протекает самопроизвольно при комнатной температуре и сопровождается заметным выделением тепла если реакция не начинается самопроизвольно, то обычно достаточно нагревания на водяной бане в течение промежутка времени от 5 мин. до нескольких дней, чтобы получить превосходные выходы гидразидов. Трудно реагирующие сложные эфиры были превращены в гидразиды путем нагревания при высокой температуре в бомбе [62, 176], но при этом может произойти декарбоксилирование, поэтому следует избегать нагревания выше 180°. Гидразиды обычно кристаллизуются при охлаждении (иногда и во время нагревания), и для получения их в чистом виде часто требуется только отделить их и высушить. Иногда образуются небольшие количества вторичных гидразидов. Отделение их не представляет трудностей, так как они нерастворимы в разбавленной кислоте и гораздо менее растворимы в органических растворителях, чем первичные гидразиды. Образование вторичных гидразидов может быть сведено к минимуму путем прикапывания сложного эфира к избытку кипящего раствора гидразингидрата с такой скоростью, чтобы не происходило никакого накопления второй жидкой фазы [11, 177, 178]. Для очистки гидразидов можно также превратить их в кристаллические изопропилиденовые производные путем нагревания с ацетоном, а затем выделить из этих производных солянокислые соли гидразидов путем обработки их в эфирном растворе сухим хлористым водородом [179]. Лишь в редких случаях очистка гидразидов производилась посредством перегонки [176] этот способ не следует применять, так как при высоких температурах, требующихся для его осуществления, 1идразиды часто вступают в реакцию конденсации, образуя гетероциклические соединения [180]. [c.348]
Безводный гидразин. Наилучший способ получения безводного гидразина заключается в перегонке гидразингидрата с твердым едким кали [265]. Хорошее описание этого способа дано в, Синтезах органических препаратов [265а]. Однако вместо указанного там прибора с пробками, обернутыми фольгой, лучше применять прибор, собранный на шлифах. Так как известны случаи бурного разложения гидразина при перегонке, ее следует проводить, закрыв прибор защитным экраном. [c.362]
Смесь ЮОг полученной таким образом соли и 20 г безводного гидразина кипятят с 75 мл абсолютного спирта в течение 1,5 час., а затем охлаждают в вакуум-эксикаторе над серной кислотой для удаления части спирта и избытка гидразина. Вещество растирают со свежей порцией абсолютного спирта, фильтруют и промывают абсолютным эфиром. Вес полученной калиевой соли моногидразида бензилмалоновой кислоты составляет 93,2 г (98,5 /q). [c.365]
Жидкий аммиак помещают в сосуд Дьюара емкостью 500 мл и небольшими порциями прибавляют к нему 60 г сернокислого гидразина, перемешивая жидкость с помощью механической мешалки. После окончания прибавления перемешивание продолжают в течение еще получаса. Затем смссь фильтруют в другой сосуд Дьюара через складчатый бумажный фильтр и дважды промывают твердый осадок жидким аммиаком для этой цели его вновь переносят в первый сосуд Дьюара, перемешивают и опять фильтруют. После испарения жидкого аммиака остается 7,5—10 г бесцветной жидкости если считать полученное вещество за безводный гидразин, то указанный выход составляет 51—68% теоретического количества. Затем к препарату прибавляют 5 мл воды. Чтобы получить гидразин-гидрат в количестве, необходимом для данного синтеза, такой опыт следует повторить три-четыре раза. [c.271]
Из химических методов определения С-концевой аминокислоты наибольшее значение имеет метод Акабори [99]. При кипячении с безводным гидразином (100 °С, 5 ч) все аминокислоты, за исключением С-концевой, превращаются в гидразиды. Отделение значительного избытка гидразидов аминокислот осуществляется реакцией с изовалериановым (или другим) альдегидом. Можно также смесь, полученную непосредственно после гидразинолиза, обработать динитрофторбензолом и после подкисления выделить ДНФ-аминокислоту. [c.368]
Получение /1-(п -метоксифенокси)бензальдегида [69]. К раствору 25,8 г неочищенного п- (л -метоксифенокси) бензгидразида в 200 мл сухого пиридина, охлажденному в бане со льдом, добавляют небольшими порциями 20 г л-толуолсульфохлорида. Смесь оставляют стоять в течение 2 час. при комнатной температуре и затем выливают в охлажденную до 0° 5 н. соляную кислоту, взятую в небольшом избытке. Выход 1-ацил-2-арилсульфонилгидра-зина составляет 34 г (88%). После двух перекристаллизаций из уксусной кислоты вещество плавится при 172—173°. 33,8 г -п-( -метоксифенокси) бензоил-2-(/г-толилсульфонил) гидразина и 150 мл этиленгликоля нагревают на металлической бане до 160°. К горячему раствору прибавляют 17,5 г безводного углекислого натрия. Наступает бурное выделение газа, которое длится около 30 сек. Нагревание продолжают течение еще 30 сек., а затем раствор быстро охлаждают примерно до 100° и разбавляют 1,2 л горячей воды. Смесь экстрагаруют эфиром эфирные вытяжки сушат сернокислым натрием и растворитель отгоняют. Кристаллический остаток представляет собой практически чистый альдегид (15,7 г, 80%). После перекристаллизации из петролейного эфира (т. кип. 60—80°) альдегид плавится при 60,5°. [c.305]
Обезвоживание гидразина. 100 г гидразинсульфата и 130 г NaOH смешивают в колбе и смесь перегоняют на масляной бане. Фракцию, полученную при 115°С и содержащую безводный гидразин, еще раз перегоняют над твердым NaOH. [c.290]
Безводный гидразин. Бе 1водпый Г. южlю получить ири выдерживании 95"(1-ного Г. в течение ночи с 20 вес. едкого калп. Полученный гель фильтруют и перегоняют фильтрат без доступа влаги 121. [c.198]
Гидразоиы. В качестве примера получения гидразонов без примесей азинов можно привести синтез гидразона ацетона [1]. Ацетон сначала превращают в азин (т. кип. 128—131°) реакцией с 100%-ным гидразингидратом и едким кали продукт затем превращают в гидразон взаимодействием с безводным гидразином и NaOH. [c.51]
Как отмечено в обзорной статье Писаревского и Мартыненко [204], по данной реакции с использованием в качестве окислителя кислорода воздуха получены формиат и ацетат висмута. Ацетат висмута получен также при использовании в качестве окислителя перекиси водорода [205]. Предложен способ получения карбоксилатов из металлического висмута, основанный на нафевании безводной смеси, содержащей карбоновую кислоту С2-С20, избыток висмута, восстановитель (гидразин) и безводный разбавитель (лифоин) при температуре 80—130 °С [206]. [c.182]
Получение гидразонов. Реакцией альдегида или кетопа с гидразином обычно не удается получить гидразон с хорошим выходом вследствие быстрого образования азина. Эту трудность можно обойти превраи1е1шем карбонильного соединения в Ы,К -диметил-гидразон с нослед ющей обменной реакцией с гидразином 141 ири кипячении Ы,Ы-диметнлгидразона с 2 3-кратиым избытком безводного гидразина [c.309]
Тип углевод-белковой связи диктует тактику получения индивидуальных олигосахаридных цепей. О-Гликозидные цепи отщепляются в условиях мягкого щелочного гидролиза в присутствии I М боргидрида натрия для предотвращения деструкции олигосахарида, при этом терминальный восстанавливающий моносахарид преврашлется а полиол. Интактные N-rликoзид вязaииыe олигосахариды удается получать лишь нагреванием (100 С, 5 ч) гликопротеина с безводным гидразином. [c.475]
Наиболее часто используемым методом дезоксигенирования насыщенных кетонов является модификация Хуанг — Минлона восстановления по Вольфу — Кижнеру [схема (113) ]. Кетон реагирует с гидразингидратом или, что менее безопасно и применяется реже, с безводным гидразином с образованием гидразона. Последний разлагают, предпочтительно in Ши, при нагревании до 150—250°С с гидроксидами натрия, калия или алкоксидом металла, полученным из высококипящего гликоля соответствующий гликоль обычно служит растворителем. Модификации позволяют вести реакцию [c.658]
Попытка получения дигидразона — дибензосуберандиона не увенчалась успехом. Мы действовали гидразингидратом или безводным гидразином на дикетон (I) или моногидразон (II) в спиртовом растворе, а также в растворе ледяной уксусной кислоты при продоллсительном нагревании. При этом реакция сопровождается сильным осмолением. Были выделены два кристаллических соединения с содержанием азота, близким к мо-ногидразону. Строения этих соединений установить не удалось. [c.109]
Полинуклеотиды. Получение апиримидиновых полимеров. Гид-разинолиз полинуклеотидов обычно проводят, используя безводный гидразин 90,9з-104 чтобы избежать расщепления фосфодиэфирных связей в щелочной среде в . Тем не менее проведение реакции с водными растворами гидразина при контролируемых значениях pH дает аналогичные результаты (см. ниже). [c.464]
Усовершенствования процесса Рашига. Как уже было указано, успех процесса Рашига зависит от скорости, с которой происходит реакция (2). Кроме того, этот процесс протекает успешно лишь в том случае, если удается предотвратить разложение гидразина хлорамином. Для этого могут быть использованы ингибиторы, способные эффективно удалять ионы металлов, которые катализируют реакции (3). Желательно также пользоваться высокими (по отношению к гипохлориту) молекулярными концентрациями аммиака. Поэтому имеет смысл указать на некоторые изменения, которые были предложены с целью усовершенствования первоначального варианта процесса Рашига. Как свидетельствует опыт, для того чтобы увеличить выход при проведении реакции в данном интервале температур, следует увеличить давление. Первоначальный процесс, предложенный Рашигом, был усовершенствован таким образом, чтобы получение гидразина протекало непрерывно. В течение всего процесса первоначальное смешивание гипохлорита и аммиака происходило при низкой температуре. Были сделаны попытки проводить работу при обычных температурах, используя очень большой избыток аммиака [108], однако выходы всегда оказывались значительно ниже теоретического. Утверждают, что выход может быть значительно увеличен [109] при следующих условиях 1)если разбавленные растворы гипохлорита и аммиака смешивать при температурах ниже 10°С, 2) если такой раствор затем полностью насытить безводным или концентрированным водным аммиаком и 3) если реакционную смесь оставить при низкой температуре до тех пор, пока не закончится образование гидразина, или до тех пор, пока не произойдет образование промежуточных продуктов, которые при слабом нагревании раствора превращаются в гидразин. Предполагается, что выход может достигать 90% теоретического. [c.36]
chem21.info
Химия гидразина
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального
образования «Челябинский государственный университет»
Химический факультет
Химия гидразина.
Курсовая работа
Выполнила студентка
группы Х-302
Воробьева Ольга
Научный руководитель:
Белик В.А.
Работа защищена
« » __________________
Оценка_________________
Челябинск, 2007 год
ВВЕДЕНИЕ
Химия гидразина изучается уже почти три четверти века. До 1875 г. были известны только симметричные дизамещенные гидразина— гидразосоединения. В 1875 г. Э. Фишер, исследуя процесс восстановления диазосоединений, выделил и охарактеризовал простые органические производные гидразина. Он получил некоторые простые арилгидразины и охарактеризовал не только свободный фенилгидразин, но также и соли этого азотистого основания. Продукты восстановления азосоединений были названы гидразосоединениями; поэтому Фишер назвал типовое вещество N2Н4 гидразином и говорил о производных этого азотоводорода как о замещенных гидразинах. Область органических производных гидразина в дальнейшем также разрабатывалась Фишером, которому в течение последующих лет удалось синтезировать моно- и дизамещенные алкил- и арилгидразины. Исследуя свойства несимметричных дизамещенных гидразинов, Фишер показал, что они могут подвергаться окислению с образованием производных тетразена, одного из гипотетических цепочечных азотоводородов.
В 1887 г. Куртиусу и его сотрудникам удалось выделить гидразин и некоторые его соли. В связи с интенсивным развитием химии гидразина (азотистого аналога этана), которое имело место в течение последующих двадцати лет, Виланд в 1913 г. писал: «Систематическому развитию химии гидразина, изученной в настоящее время, достаточно полно, в значительной мере способствовало открытие вещества. Практически все комбинации гидразина с различными типами органических соединений могли быть затем получены из этого весьма реакционноспособного вещества. Производные гидразина вскоре превысили по числу и многообразию органические производные аммиака, которые изучались в течение многих десятилетий».
Гидразин является одним из простейших азотоводородов. Если сравнить этот класс соединений с углеводородами, то гидразин можно рассматривать как аналог этана. Он является вторым азотоводородом, выделенным в свободном состоянии. До настоящего времени в свободном состоянии было получено всего лишь три азотоводорода, а именно аммиак, гидразин и азид водорода. Другие азотоводороды давно известны в виде органических производных, и многие из них могут быть получены только в результате окисления соответствующих производных гидразина.
1.ПОЛУЧЕНИЕ ГИДРАЗИНА
Гидразин впервые был получен в виде органических производных. В 1887 г. Куртиус синтезировал и выделил неорганические соли, а также гидрат гидразина. Первые методы синтеза солей гидразина, из которых удалось получить его гидрат, были основаны главным образом на восстановлении соединений, содержащих связь азот—азот. Лишь позднее были предприняты попытки использовать в качестве исходного вещества аммиак и получать гидразин путем разложения или окисления аммиака и его производных.
Реакции, которые приводят к образованию гидразина, практического применения не получили. Синтез Рашига, включающий в себя частичное «окисление» аммиака (и мочевины) гипохлоритом, является единственным препаративным методом, который применяется для получения гидразина в производственных масштабах.
1.1 Восстановление соединений, содержащих связь азот-азот
Теоретически любое соединение, содержащее связь азот—азот, может быть восстановлено до гидразина или его производного. Соответствующие методы получили широкое применение в органической химии для получения органических производных гидразина; некоторые из них в результате последующей обработки дают соли гидразина или сам гидразин. Так, например, гидразин был получен из азотноватистой кислоты и ее изомеров, нитрамида и нитрозо-гидроксиламина, из бимолекулярных нитрозосоединений, а также из нитрозоаминов, азосоединений и азидов. Кроме того, в качестве исходных веществ были использованы нитриты, нитраты и другие нитросоединения, однако восстановление их, вероятно, протекает с образованием промежуточных соединений, содержащих связь азот—азот. Утверждали даже, что при некоторых условиях молекулярный азот может реагировать с водородом, образуя гидразин; следовательно, вполне возможно, хотя и мало вероятно, что, изменив условия, используемые в процессе синтеза аммиака, можно получить гидразин. Однако ни один из этих методов не послужил основой для промышленного способа получения гидразина, главным образом вследствие того, что для практических целей наблюдающиеся выходы слишком низки.
Получение из нитрамида ( NH 2 NO 2).
Нитрамид, являющийся несколько более устойчивым изомером азотноватистой кислоты, может быть восстановлен до гидразина в кислом растворе цинковой пылью. Большой интерес представляют методы, включающие
восстановление ацилпроизводных нитрамида, в особенности таких соединений, как нитромочевина и нитрогуанидин, поскольку не исключено, что эти методы могуг быть использованы в промышленности. Указанные вещества восстанавливаются до соответствующих производных гидразина, а именно до семикарбазида и аминогуанидина, в результате гидролиза которых может быть получен гидразин. Nh3NO2N2Н4Nh3CONHNO2
+6[Н] Nh3СОN2Н3+2Н2ONh3С(NH)NHNO2 Nh3С(NH)(N2Н3)
Получение из азотоводородной кислоты и азидов
Гидразин также был получен восстановлением азидной группы. Восстановление может протекать как в щелочном, так и в кислом растворе под действием различных химических агентов, например амальгамы натрия, цинка и соляной или серной кислоты, сульфида натрия или гидроокиси двухвалентного железа. Эти реагенты дают главным образом аммиак и азот и только в очень небольших количествах гидразин. Большой выход получается только в том случае, если гидразин немедленно удаляется из сферы реакции при помощи некоторых методов, например осаждения в форме такой слабо растворимой двойной соли, как сульфат цинка — сульфат гидразина. При восстановлении азотоводородной кислоты в кислом растворе цинком в отсутствие катализатора получаются тем большие выходы гидразина, чем выше температура.
В результате электролиза водных растворов азидов с использованием различных металлических анодов образуется главным образом азот наряду с некоторым количеством аммиака и гидразина. Выходы относительно малы.
Рассмотрим различные стороны реакции Шмидта. Азотоводородная кислота и азиды служат источником имидного радикала, который может реагировать со вторым таким же радикалом, как растворителя, так и растворенных веществ.
Nh4 → [NH] + h3.
Если раствор азида водорода в бензоле оставить стоять с концентрированной серной кислотой при 15°С, то наряду с такими соединениями, как анилин и гидроксиламин, в сернокислотном слое образуется некоторое количество гидразина.
Выход гидразина заметно понижается с повышением температуры. При 60°С получается только анилин. Было доказано, что имидный радикал, соединяясь с другим таким же радикалом, образует диимид, который затем диспропорционируется, причем получаются азот и гидразин.
2[НN]→N2Н2; 2[N2Н2]→N2Н4 + N2.
Вероятность такого механизма следует из того факта, что калиевая соль азодикарбоксиловой кислоты подвергается гидролизу в кислом растворе, образуя азот и гидразин, а также азотоводородную кислоту, окись углерода и аммиак. В некотором отношении эту реакцию можно рассматривать как процесс самоокисления производного диимида.
Сульфат гидразина был получен при поглощении безводного азида водорода концентрированной серной кислотой. Механизм этой реакции не был точно выяснен; однако и в этом случае можно сделать предположение о промежуточном образовании радикала NH.
1.2 Разложение аммиака
При полном разложении аммиака образуются водород и азот. Однако было точно установлено, что эти вещества не получаются на первой стадии процесса разложения. Экспериментально была показана возможность побочных реакций с образованием промежуточных продуктов, одним из которых является гидразин. По мнению Рашига, первая стадия разложения аммиака протекает с образованием радикала NН. Можно предположить, что этот радикал способен далее вступать в реакцию конденсации с аммиаком, в соответствии со следующим уравнением:
N3 — NH+2[Н],
NН+NН3→ [НNNН3] → N2Н4.
Cчитают, что одним из промежуточных продуктов разложения аммиака является радикал NН2 и что он может соединяться со вторым таким же радикалом, образуя гидразин.
Nh4 → Nh3 + [Н]; Н2N+Nh3 → N2Н4.
Существенным является то обстоятельство, что экспериментальные методы и условия, используемые при разложении аммиака, приводят к еще более быстрому разложению гидразина. Для того чтобы экспериментально продемонстрировать образование гидразина как промежуточного или побочного продукта, необходимо немедленно удалить гидразин из сферы реакции или быстро охладить это соединение, чтобы повысить его устойчивость. Разложение аммиака было осуществлено путем пиролиза, фотохимически в результате фотосенсибилизации, действием электрического разряда на газообразный аммиак и при бомбардировке электронами.
mirznanii.com
| Получение золота чистоты 0,999 в домашних условиях
Сначала немного ликбеза. Золото, которое мы привыкли видеть в виде ювелирных изделий представляет собой сплавы. Например, обычное жёлтое золото 585 пробы - это сплав, содержащий в 1 грамме 585мг(58,5%) золота, 10% серебра, до 1% металлов платиновой группы, 2%-кадмий, цинк, хром, др., остальное - медь. Белое золото отличается от жёлтого содержанием никеля и хрома за счёт уменьшения доли меди. Чтобы получить из более высокопробного золота золото с низшей пробой достаточно добавить в сплав медь (золото приобретет красноватый оттенок). Для повышения пробы требуются некоторые химические реакции. В промышленных и лабораторных условиях очистка золота сопряжена с применением высокотоксиных реагентов(например, ртути, цианидов). Это необходимо при очищении природного (самородного) золота от свинца, металлов платиновой группы, др.. Ювелирное золото(которое в изделиях) уже не требует столь сложных способов очистки. Убрать медь, хром, никель и др. неинертные(неблагородные) металлы можно с помощью простой ионнообменной реакции: помещаете золото в 95-96% азотную кислоту(желательна вытяжка), лучше, если золото предварительно измельчено, например, напильником. Температуру раствора желательно иметь больше 20С. При добавлении (осторожно, мелкими порциями) к раствору чистого цинка происходит растворение сплава золота, растворение цинка и выделение из раствора сплава золота 900-916 пробы, содержащего золото, серебро и металлы платиновой группы. Золото осаждается хлопьями, которые следует извлечь из раствора путем фильтрования и промывания водой.Затем при помощи газовой или бензиновой горелки(1200С) золото переводят в металический вид.Это так называемое "зубное золото", используемое в стоматологии и имеющее большую мягкость и более жёлтый цвет. Чтобы получить абсолютно чистое золото пробы 0,999 следует взять ювелирное золото любой пробы, растворить его в "царской водке", которая получается смешиванием концетрированных 1 части азотной и 2-х частей соляной кислот. Осторожно!!! Нужна термостойкая хим.посуда и вытяжка!! При растворении золота раствор нагревается, выделяются пары кислот и токсичный оксид азота IV. И этой реакции получился раствор хлорного золота. В растворе также по-прежнему остаются все остальные металлы. Далее. Берем порошок хлорного железа, делаем насыщенный водный раствор хлорного железа и титруя, покапельно добавляем в содержащую золото царскую водку. Хлопьями осаждается серебро. Удаляем серебро фильтрованием и промыванием в воде(если оно вам нужно, то горелкой превращаем его в металлическую форму). Выделяем само золото. Берем гидразин. Гидрозин это белый порошок без запаха, абсолютно нетоксичен, в отличие от всем известного триметилгидразина-компонента гептила, который входит в состав ракетного топлива. Создаем насыщенный водный раствор гидразина и титруя, покапельно добавляем в раствор царской водки, из которой мы уже удалили серебро. Хлопьями коричневого цвета выделяется чистое золото. После аккуратного удаления хлопьев золота из раствора их следует перевести в металлический вид горелкой не менее 1200С, Сплавление производят в асбестовом или керамическом тигеле. Желательно добавить флюс - буру(натриевая соль лимонной кислоты). Получившееся золото имеет чистоту 0,999, идентичную золоту банковских слитков. Оно имеет выраженно жёлтый цвет -"цвет дешевой бижютерии". Из него можно делать слитки, белое и жёлтое золото любых проб, золотые припои для пайки золотых изделий. Таблицы процентных содержаний металлов в различных видах и пробах золота, золотых припоев вы найдёте в любом руководстве по ювелирному делу. Все приведенные выше реактивы продаются свободно в магазинах, торгующих хим. реактивами, самый дорогой среди них-гидразин 2000 руб/кг., остальные на порядок дешевле. Бура продается также и в аптеках.
|
www.irkidei.ru
Гидразин - Получение
Химия - Гидразин - Получение
01 марта 2011Оглавление:1. Гидразин2. Получение3. Токсичность
Гидразин получают окислением аммиака Nh4 или мочевины CO2 гипохлоритом натрия NaClO:
Nh4 + NaClO Nh3Cl + NaOH Nh3Cl + Nh4 N2h5 · HCl,
Nh3Cl + NaOH Nh3Cl + Nh4 N2h5 · HCl, реакция проводится при температуре 160 °C и давлении 2,5−3,0 МПа.
Синтез гидразина окислением мочевины гипохлоритом по механизму аналогичен синтезу аминов из амидов по Гофману:
h3NCONh3 + NaOCl + 2 NaOH N2h5 + h3O + NaCl + Na2CO3, реакция проводится при температуре ~100 °C и атмосферном давлении.
Гидразин как топливо
Гидразин и его производные, такие как НДМГ и Аэрозин широко распространены как ракетные горючие. Они могут быть использованы как в паре с окислителем, так и в качестве однокомпонентного топлива в данном случае рабочим телом двигателя являются продукты разложения на катализаторе. Последнее удобно для маломощных двигателей.Во время Второй мировой войны гидразин был применён в Германии на реактивных истребителях «Мессершмитт Ме-16З».
Теоретические характеристики различных видов ракетного топлива, образованных гидразином с различными окислителями.
| Фтор | 364,4 с | °С | 1,314 | 5197 м/с | 31 % |
| Тетрафторгидразин | 334,7 с | °С | 1,105 | 4346 м/с | 23,5 % |
| ClF3 | 294,6 с | °С | 1,507 | 4509 м/с | 27 % |
| ClF5 | 312,0 с | °С | 1,458 | 4697 м/с | 26,93 % |
| Перхлорилфторид | 295,3 с | °С | 1,327 | 4233 м/с | 40 % |
| Фторид кислорода | 345,9 с | °С | 1,263 | 4830 м/с | 40 % |
| Кислород | 312,9 с | °С | 1,065 | 3980 м/с | 52 % |
| Перекись водорода | 286,9 с | °С | 1,261 | 4003 м/с | 33 % |
| N2O4 | 291,1 с | °С | 1,217 | 3985 м/с | 43 % |
| Азотная кислота | 279,1 с | °С | 1,254 | 3883 м/с | 40 % |
- Удельная тяга равна отношению тяги к весовому расходу топлива; в этом случае она измеряется в секундах. Для перевода весовой удельной тяги в массовую её надо умножить на ускорение свободного падения
Гидразин в топливных элементах
Гидразин широко применяется в качестве топлива в гидразин-воздушных низкотемпературных топливных элементах.
Гидразин в производстве взрывчатых веществ
Нитрат и перхлорат гидразина применяются в качестве очень мощных взрывчатых веществ разных сортов астролита. Они обладают большой скоростью детонации. Жидкая смесь гидразина и нитрата аммония используется как мощное взрывчатое средство с нулевым кислородным балансом.
Просмотров: 4886
4108.ru







Поиск по сайту
Email рассылка
Узнавай первым
об обновлениях на сайте по Email БЕСПЛАТНО! Как только на сайте появятся новые посты, видео или фото, Ты сразу же будешь извещен об этом одним из первых.
Новое на сайте
Новое на форуме
Нет сообщений для показа
